
Chemistry Development Kit (CDK)
The Steinbeck group founded and now co-develops the Chemistry Development Kit (CDK), the leading open-source Java library for structural chemo- and bioinformatics. The CDK covers a wide range of functionality needed for performing virtual compound screening, property prediction and many other tasks of molecular informatics. In addition to its virtues for developing open systems in structural bioinformatics, it is a valuable tool for teaching. With 90.000 non commenting code statements (NCSS) in over 9000 methods in 900 classes, the CDK provides a basis for studying hands-on examples for the standard algorithms used in handling and modifying molecular structures as well as for calculating their properties, written in a modern object-oriented language, using commonly accepted design patterns.
Natural-Product-Likeness
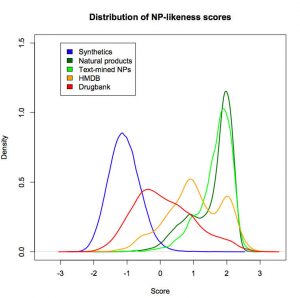 Natural product-likeness of a molecule, i.e. similarity of this molecule to the structure space covered by natural products, is a useful criterion in screening compound libraries and in designing new lead compounds. A closed source implementation of a natural product-likeness score, that finds its application in virtual screening, library design and compound selection, has been previously reported by Peter Ertel. We then worked with him to product an open-source and open-data re-implementation of this scoring system and illustrated its efficiency in ranking small molecules for natural product likeness.
Natural product-likeness of a molecule, i.e. similarity of this molecule to the structure space covered by natural products, is a useful criterion in screening compound libraries and in designing new lead compounds. A closed source implementation of a natural product-likeness score, that finds its application in virtual screening, library design and compound selection, has been previously reported by Peter Ertel. We then worked with him to product an open-source and open-data re-implementation of this scoring system and illustrated its efficiency in ranking small molecules for natural product likeness.
The NP-Likeness scorer now lives at https://github.com/CS76/NP-Likeness.
A workflow-based version of the Natural-Product-Likeness scoring system is implemented as Taverna 2.2 workflows, and is available under Creative Commons Attribution-Share Alike 3.0 Unported License athttp://www.myexperiment.org/packs/183.html. It is also available for download as executable standalone java package fromhttp://sourceforge.net/projects/np-likeness/under Academic Free License.
Our open-source, open-data Natural-Product-Likeness scoring system can be used as a filter for metabolites in Computer Assisted Structure Elucidation or to select natural-product-like molecules from molecular libraries for the use as leads in drug discovery.
Bioclipse
 In collaboration with Jarl Wikberg’s group and now Ola Spjuth’s group at the University of Upsala, Sweden, we co-founded the Bioclipse project to build a plug-in based, rich client desktop workbench for molecular informatics. Bioclipse won the JAX conference audience award for important European contribution to the development of Eclipse in 2006. On November 2007, the project was recognised with a jury prize in the 4th edition of the Trophées du Libre.
In collaboration with Jarl Wikberg’s group and now Ola Spjuth’s group at the University of Upsala, Sweden, we co-founded the Bioclipse project to build a plug-in based, rich client desktop workbench for molecular informatics. Bioclipse won the JAX conference audience award for important European contribution to the development of Eclipse in 2006. On November 2007, the project was recognised with a jury prize in the 4th edition of the Trophées du Libre.
Our group contributed plug-ins for spectrum handling, database editing and extension of Bioclipse’s Systems Biology capabilities. The spectrum facilities are grouped in the Speclipse feature. The integration of an Systems Biology Markup Language (SBML) editor and the integration of metabolomics simulations will be the next step. Bioclipse is a state-of-the-art, user-friendly, open-desktop application for performing System Biology Simulations.
JCAMP-DX
The JCAMP-DX project is the reference implemention of the IUPAC JCAMP-DX spectroscopy data standard. It implements a parser and a writer to convert JCAMP-DX files to Java objects.
Historic Archive
A number of application which we developed in the past are now outdated for various reasons or are simply not developed by us anymore. They are listed here for documentation purposes.
NMRShiftDB
Nmrshiftdb was a web-based database for organic structures and their nuclear magnetic resonance (nmr) spectra. It was originally developed in our group by Stefan Kuhn and funded by the German Research Council (DFG). Stefan Kuhn now thankfully continues to develop the database under the name NMRShiftDB2, which is now hosted by our colleague Niels Schloerer at the Univeristy of Cologne. NMRShiftDB2 allows for spectrum prediction (13C, 1H and other nuclei) as well as for searching spectra, structures and other properties. Last not least, it features peer-reviewed submission of datasets by its users. The nmrshiftdb2 software is open source, the data is published under an open content license.
JChemPaint
JChemPaint (JCP) is an editor and viewer for 2D chemical structures developed using CDK. It is implemented in several forms: a Java application and two varieties of Java applet.
Pros:
It can also be used as a component to embed in other applications. JCP has well tested an user-friendly interface. Its behaviour is consistent in the application and the applet.
JChemPaint offers:
- Drawing and deletion of single, double, triple and stereo bonds
- Ring templates (3-8 atoms) with one-click attachment
- An extensive template library
- Colouring of atom types, and other rendering settings
- Editing of atomic charges, isotopes and hydrogen count
- Loading and saving of structures in Chemical Markup Language (CML) and as MDL MOL files and SDF files (loading only)
- Automated Structure Layout, also known as Structure Diagram Generation
- Loading structures from the Internet using CAS or NSC number
- Normalisation of structures, currently limited to aromaticity detection
- Saving bitmap pictures of the structures
- Saving structures as graphics (PNG, BMP, Scalable Vector Graphics (SVG))
- Postscript printing
- The amount of novel basic research to be performed while developing the CDK goes significantly beyond what is to be expected from what looks like a pure infrastructure project. Questions of how to perceive aromaticity, perform fingerprinting of structures or define pharmacophore queries, are often researched and published in CDK context for the first time.
Cons:
It is an applet. The web is migrating away from applets, towards Javascript-based interactivity of web components. There are now free, lightweight, open-source structure editors written in Javascript, like Ketcher, which one can use.
OrChem
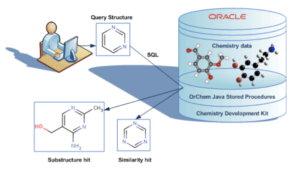 OrChem is an Oracle database chemistry plug-in using the CDK. For chemistry databases, various commercial “cartridges” exist that facilitate searching and analyzing chemical data. OrChem also provides functionality like this, but is not a cartridge. It doesn’t need Oracle’s extensibility architecture because its Java components run as Java stored procedures inside the Oracle standard JVM (Aurora).
OrChem is an Oracle database chemistry plug-in using the CDK. For chemistry databases, various commercial “cartridges” exist that facilitate searching and analyzing chemical data. OrChem also provides functionality like this, but is not a cartridge. It doesn’t need Oracle’s extensibility architecture because its Java components run as Java stored procedures inside the Oracle standard JVM (Aurora).